AlphaFold 2 paper and code is finally released. This post aims to inspire new generations of Machine Learning (ML) engineers to focus on foundational biological problems.
This post is a collection of core concepts to finally grasp AlphaFold2-like stuff. Our goal is to make this blog post as self-complete as possible in terms of biology. Thus in this article, you will learn about:
The central dogma of biology
Proteins and protein levels
Amino acids, nucleotides and codons
Protein structure characteristics such as Domains, Motifs, Residues and Turns
Distograms
Phenotypes and genotypes
Multiple sequence alignment
Biology tasks that can be approached with ML
Association of biology and ML model design
We assume that you have no background in biology and a bit of background in ML.
Before anything let’s put ourselves in some broader context. A great introduction that I chose is Ken Dill’s talk at TEDx:
This is pretty much where the scientific community was in 2013. Who could possibly imagine deep learning would penetrate such a niche field! And even further, to create the largest public database of proteins:
AlphaFold’s Protein Structure Database provides open access to protein structure predictions for the human proteome and 20 other organisms to accelerate scientific research. At the time of writing this article, there exist 350K proteins and they are planning to expand it to every protein known to humans (almost 100M)!
If that triggers your curiosity, hop in for something completely different!
Minimal biology prerequisites
Wondering where to start? Start from DNA and RNA to get a hold of the central dogma of biology!
DNA
DNA is a molecule composed of two complementary chains that coil around each other to form a double helix. The chains are also called strands in the literature. Complementary DNA strands are antiparallel to each other. Imagine that the DNA “elements” at each position are mirrored from one strand to the other. This pairing allows cells to copy information from one generation to another and even fix errors in the information stored in the sequences. The DNA molecule carries genetic instructions for the development, functioning, growth and reproduction of the organism.
 Source: Alberts B, Johnson A, Lewis J, et al., Molecular Biology of the Cell (2002)
Source: Alberts B, Johnson A, Lewis J, et al., Molecular Biology of the Cell (2002)
RNA
RNA is a single-stranded molecule that is essential in coding, decoding, regulating and expressing genes. Out of many types of RNA, the three most well-known and most commonly studied are messenger RNA (mRNA), transfer RNA (tRNA), and ribosomal RNA (rRNA), which are present in all organisms. In protein expression, we are particularly interested in mRNA, which acts as a portable transcript, of the instructions written in genes, to ribosomes, the cell's machinery responsible for producing a protein.
The central dogma of biology
Essentially, the central dogma of biology explains the flow of genetic information in a biological system and its forward path consists of three steps:
The DNA is replicated (inside the nucleus). A DNA cell makes an identical copy of its genome before it divides itself into two separate chains.
The DNA chain is used as a template to produce a complementary RNA copy in a process called transcription. This will again happen inside the nucleus.
The RNA chain is decoded and translated by ribosomes to produce a polypeptide sequence, otherwise known as a protein. This process is called translation and is happening outside of the nucleus.

The 4 fundamental DNA elements, called bases, are {A, T, C, G} and they will be our sequence elements later on! RNA consists of 4 bases as well but T is replaced with U: {A, U, C, G}. Every DNA or RNA sequence is formulated as a combination of these 4 bases.
The following animation can illustrate the process in great detail:
Proteins, amino acids, nucleotides and codons
By now you get the idea that cells generate proteins, which are sequences of amino acids. A polypeptide is a polymer of amino acids joined together by peptide bonds. In other words, amino acids are the structural elements of all proteins. There are many known amino acids but only 20 are encoded by the universal genetic code. Genetic code refers to all the valid combinations of nucleotide bases. Specifically, 3 adjacent nucleotides constitute a unit known as the codon, which codes for an amino acid.
For example, the sequence AUG (in the mRNA) is a codon that specifies the amino acid methionine, which almost always specifies the beginning of a protein. There are 64 possible codons. Out of them, there are 3 codons that do not code for amino acids but indicate the end of a protein. The remaining 61 codons specify the 20 amino acids that make up proteins. Below is the table that demonstrates the valid combinations.

The following video clarifies many aspects on proteins:
We find it very important to highlight the different visualization of the proteins, as described in the video:

If you are curious to know all the amino acids names and symbols, consult this table.
The 4 levels of protein structures
To describe a protein’s structure, we use 4 distinct levels. The following figure sums up the protein structural levels from a biological standpoint:
 Source: Figure after © 2010 PJ Russell, iGenetics 3rd ed.; all text material © 2014 by Steven M. Carr
Source: Figure after © 2010 PJ Russell, iGenetics 3rd ed.; all text material © 2014 by Steven M. Carr
1) The primary structure is simply the sequence of amino acids in a polypeptide chain.
 The hormone cow insulin has two polypeptide chains, A and B, shown in diagram below. Each chain has its own set of amino acids, assembled in a particular order. Source and Image credit: OpenStax Biology.
The hormone cow insulin has two polypeptide chains, A and B, shown in diagram below. Each chain has its own set of amino acids, assembled in a particular order. Source and Image credit: OpenStax Biology.
2) In very simple terms, secondary structure refers to the 3D arrangement of stable local repeating structures. A stricter definition defines it as regular, recurring 3D arrangements of adjacent amino acids. The gist here is the two secondary structures: the α-helix (shown here) and the β-structures (aka beta sheets).
 Alpha helices vs beta sheets. Source: bioninja.com
Alpha helices vs beta sheets. Source: bioninja.com
3) The tertiary structure is the overall 3-D shape of the protein molecule. It is used to describe the spatial relationship of the secondary structures to one another.
4) Quaternary describes protein-protein interactions in closely packed arrangements. In other words, when many polypeptide chains gather together to form a functional molecule (usually called protein subunit). Note that not all proteins have a quaternary structure.
 An example of a protein with a quaternary structure is haemoglobin (O2 carrying molecule in red blood cells)Source: bioninja.com
An example of a protein with a quaternary structure is haemoglobin (O2 carrying molecule in red blood cells)Source: bioninja.com
Left: tertiary structure of a single polypeptide. Right: quaternary structure of Haemoglobin (the oxygen-carrying molecule in red blood cells), composed of four (polypeptide) chains. Notice that the tertiary 3D structure is not altered.
A closer look at protein folding: Domains, Motifs, Residues and Turns
There is some niche terminology that would be beneficial to clarify before you jump in any bioinformatic project:
Domains
Domains: A (structural) domain is a self-stabilizing part of the protein's polypeptide chain. It often folds independently of the rest of the protein chain (they will not unfold if separated). Furthermore, domains are responsible for a specific self-contained job in the protein. Like the illustrated pink domain below, domains are not unique to the protein. Instead, molecular evolution uses domains as building blocks, recombining them in different arrangements, but with identical folding, to create proteins with different functions.
 The structures of two different proteins shown below share a common PH (Pleckstrin Homology) domain (maroon). Source: LibreTexts:Protein Domains, Motifs, and Folds in Protein Structure
The structures of two different proteins shown below share a common PH (Pleckstrin Homology) domain (maroon). Source: LibreTexts:Protein Domains, Motifs, and Folds in Protein Structure
Structural motifs
Structural motifs: small structure-dependent 3D regions formed from connecting different secondary structural elements (e.g. α-helices and β-sheets). They are also shared among different proteins. However, they are bound to the protein with multiple hydrogen bonds that influence its shape, which makes their folding dependent on the overall 3D structure of the protein.
Thus, motifs do not retain their function when separated from the larger protein they are part of. In other words, motifs lose their function due to the loss of hydrogen bonds. When separated, they unfold because they lose their structure and, as a result, their function.
 Commonly observed motifs Source:Principles of Biochemistry
Commonly observed motifs Source:Principles of Biochemistry
There are certain motifs that occur over and over again in different proteins. The helix-loop-helix motif, for example, consists of two α-helices joined by a reverse turn. The Greek key motif consists of four antiparallel β-strands in a β-sheet where the order of the strands along the polypeptide chain is 4, 1, 2, 3. The β-sandwich is two layers of β-sheets.
Finally, note that there can be motifs on the primary structure level. In this case, a motif describes a consensus sequence of amino acids. A common motif may adopt similar conformations in different proteins.
 Source: Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th edition.
Source: Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th edition.
Similar sequences can adopt alternative conformations in different proteins. Here, the sequence VDLLKN (in red) assumes an α-helix in one protein context (left) and a β-strand in another (right).
Residues and side chains
A residue is a single molecular unit within a polypeptide. Simply, it’s just another term for amino acids. The three building blocks of an amino acid are 1) amine group, 2) a carboxylic acid group, and 3) the R-group or side chain.
Sidechains are the variable part of amino acids.
They influence the features of amino acid, such as how it interacts with water, help guide the structure of a finished protein, as well as protein-protein interactions.
When dealing with protein datasets, note that each amino acid has both a one-letter and three-letter abbreviation.
 The defining feature of an amino acid is its side chain (at top, blue circle; below, all colored circles). Source:Nature Education, 2010.
The defining feature of an amino acid is its side chain (at top, blue circle; below, all colored circles). Source:Nature Education, 2010.
Gist: The defining feature of an amino acid is its side chain, as shown on the upper part of the figure (blue circle); or on the lower part with coloured circles. When connected together by a series of peptide bonds, amino acids form a polypeptide. The polypeptide will then fold into a specific conformation depending on the interactions (dashed lines) between its amino acid side chains.
Some amino acid residues are polar, meaning they have a charge. These polar amino acid residues are hydrophilic, meaning they interact with water, or hydrophobic. In protein folding, hydrophilic residues are exposed to water and hydrophobic residues are hidden from water.
Turns and loops
Turns and loops are types of secondary protein structures that connect α-helices and β-strands.
They usually cause a change in direction of the polypeptide chain allowing it to fold back on itself to create a more compact structure.
Loops generally have hydrophilic residues. They can be found on the surface of a protein. Loops that contain 4 or 5 amino acid residues are called turns. Turns are well-defined structural elements and are considered as the third form of secondary structure (along with the α-helix and β-strand). The most common types being type I and II β-turns.
 Example of motifs. Left: A β-hairpin motif. Right: Helix-Turn-Helix (HTH) motif.
Example of motifs. Left: A β-hairpin motif. Right: Helix-Turn-Helix (HTH) motif.
Left: A β-hairpin motif consists of two strands that are adjacent in primary structure, oriented in an antiparallel direction, and linked by a short loop of two to five amino acids (green). Right: Helix-Turn-Helix (HTH), a major structural motif capable of binding DNA (right). HTH incorporates two α-helices, joined by a short strand of amino acids (light blue), that bind to the major groove of DNA. The HTH motif occurs in many proteins that regulate gene expression.
You are probably wondering how we represent such complex 3D structures. This brings us to distograms.
What is a distogram?
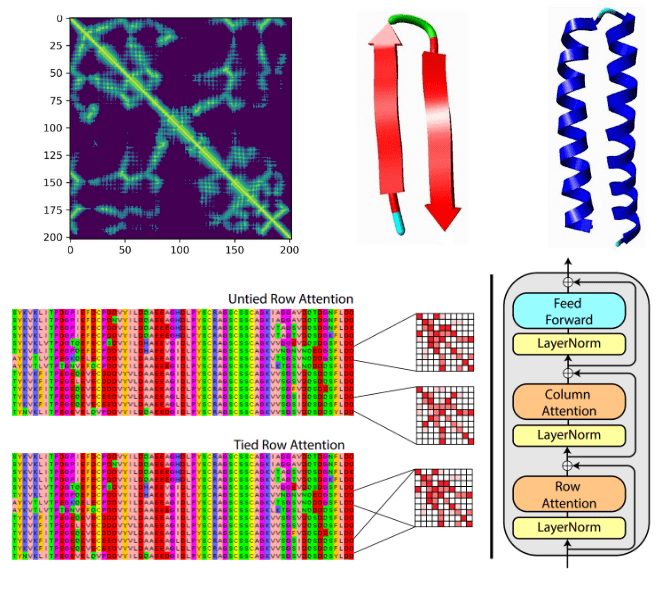
The distogram is the key intermediate step to protein folding. For a sequence of length , a distogram (distance + histogram) is an matrix, which shows the histogram of the pairwise distances. The distances are “binned” so it is regarded as a binned distance distribution. If the distances are binned the distogram may have as many channels as bins meaning tensor. Distograms are also called contact maps and they are always symmetric matrices.
 Source:Deep Learning-Based Advances in Protein Structure Prediction, Pakhrin et al 2021
Source:Deep Learning-Based Advances in Protein Structure Prediction, Pakhrin et al 2021
Based on the paper “Deep Learning-Based Advances in Protein Structure Prediction” by Subash C. Pakhrin et al.:
“The problem of protein distogram or real-valued distance prediction (bottom row) is similar to the depth prediction problem in computer vision (top row). In all these problems, the input to the Deep Learning model is a volume (3D tensor). In computer vision, 2D images expand as a volume because of the RGB channels. Similarly, in the case of distance prediction, predicted 1D and 2D features are transformed and packed into 3D volume with many channels of inter-residue information” ~ Subash C. Pakhrin et al. 2021
But why distograms? Well, the distances in a distogram are relative, meaning that the inter-residue distances are invariant to 3D rotations and translations. This makes the task much simpler.
Genotype VS phenotype
Last but not least, you have to distinguish between genotype (the encoded info) vs phenotype (what can be observed):
In a nutshell, an organism’s genotype is the set of genes that it carries while the phenotype is all of its observable characteristics; which are influenced both by its genotype and by the environment.
It is very common for the phenotype to be our target in a supervised machine learning setup!
Biological tasks that can be approached with ML
MSA (Multiple sequence alignment)
I kinda freaked out when I saw “MSA” in some discussion groups about Alpha Fold. So let’s start with this:
Multiple sequence alignment (MSA) refers to the process or the result of sequence alignment of three or more biological sequences, generally a protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor.
Below is an illustration of aligned sequences:

So, MSA are aligned sequences with strong structural (evolutional) similarities. Alpha fold heavily relies on MSA as an additional input. Instead of the particular primary structure of the protein (target), it finds multiple similar proteins and aligns them together. This process can be regarded as additional information for the model that will work like hand-crafted features.
Why is this useful?
Because the 3D structure is more conserved than the primary sequence structure. The sequence can be slightly altered but the overall structure that is related to protein function is preserved! MSA provides a hint towards this path and it’s one of the main reasons that deep learning methods emerged and shined in protein folding.
How can you find similar sequences?
Well, one way is by searching large datasets of protein sequences derived from DNA sequences and aligning them to the target sequence to generate an MSA. The alignment can happen at the gene, protein, and metagenomics levels. Various methods are used within MSA to maximize scores and the correctness of alignments. Each method uses a heuristic that tries to replicate the evolutionary process and get a realistic alignment. The three examples below have been used by the DeepMind team, with JackHMMER, a Hidden Markov Models approach, appearing in both AlphaFold 1 and 2.
Correlated changes in the positions of two amino acid residues across MSA sequences can be used to infer which residues might be in contact.
We refer to the above as evolutionary covariation.
Protein 3D structure prediction
Wondering why Alpha Fold is so important? Because it tackles a very crucial task of human existence: protein structure prediction. Below we found a beautiful illustration of a protein that folds to shape it’s final 3D structure:
The 3D structure determines the protein’s function (how it works). Predict the structure and you know the functionality.
Formally, protein structure prediction is the inference of the 3D structure of a protein from its amino acid sequence (input). In biological terms, the 3D structure is the secondary and tertiary structure (output). ~ Wikipedia
One can formulate the problem as a 3D contact prediction. Here is a ribbon diagram of the desired distances measured in Angstroms.
 Two globular proteins with some contacts in them shown in black dotted lines along with the contact distance in Armstrong. The alpha helical protein 1bkr (left) has many long-range contacts and the beta sheet protein 1c9o (right) has more short- and medium-range contacts. Source: Protein Residue Contacts and Prediction Methods
Two globular proteins with some contacts in them shown in black dotted lines along with the contact distance in Armstrong. The alpha helical protein 1bkr (left) has many long-range contacts and the beta sheet protein 1c9o (right) has more short- and medium-range contacts. Source: Protein Residue Contacts and Prediction Methods
The alpha-helical protein (left) has many long-range interactions while the beta-sheet protein (right) has mostly medium-range interactions.
Genotype to phenotype prediction
Another super common task is predicting phenotypes from genotypes. Below is an example of a genotype-to-phenotype task, where computer vision and NLP methods are used. The aim is to both predict and classify toehold switch performance (phenotype) from RNA sequences (genotype).

Toehold switches are RNA molecules of increased interest because they act as programmable sensors for precision diagnostics. Note that one-hot encoding turns RNA into a tensor, where is the length of the RNA sequence. For the prediction task, convolutional neural networks are used. On the other hand, the RNA sequences are tokenized into 3-mers (triplets) and fed into a recurrent neural network for binary classification.
What kind of bioinformatic datasets are we dealing with?
In the majority of cases, we have a labelled dataset, with sequences as inputs. The inputs might be DNA, RNA, protein, or amino acid sequences. The length of these sequences is arbitrary. We may also have some attributes that correspond to their properties (i.e. thermodynamic properties).
Ok, I said labelled data. So what’s our target?
The target we are trying to predict is either one of the phenotypes (in a classification or regression task) or the entire 3D structure of a protein as in AlphaFold 2.
Representing DNA and amino acid sequences
Let’s inspect how we can represent biological sequences. And to do that we need to understand what our tokens are.
We encode spoken language at a word-level and each input sequence is a sentence. In the DNA world, we have a character-level encoding. Intuitively, you can imagine each DNA seq. to be a word and each token being a character. However, instead of a dictionary of characters from A to Z, we have only 4 basic elements (bases): {Α, Τ, G, C} for DNA and {Α, Τ, G, U} for RNA. This is our nice little dictionary.
In amino acids, we have 20 elements in our dictionary, as there are only 20 amino acids that can be produced from the human genome.
A different approach is to use n-gram-like representations (sequence of consecutive things), namely k-mers. We define k-mers as overlapping or non-overlapping, substrings of length k contained within a biological sequence. For example, for the DNA sequence TAGACTGTC, we get five possible overlapping 5-mers: {TAGAC, AGACT, GACTG, ACTGT, CTGTC}. In this way, we create a different form of embedding, where our vectors contain the number each k-mer occurs for a given sequence! k-mer encoding is a simple representation strategy when dealing with datasets consisting of samples with varying lengths.
Association of biology with ML model design
As with everything else, ML modelling requires domain knowledge. To this end, I will present some examples.
Attention mechanism for processing MSA
Protein structure prediction can be focused on drug & molecule design. A very recent example is the MSA Transformer that modifies the attention mechanism. It is an excellent example of where ML theory and domain knowledge come together.
Based on tied row attention, they trained a large unsupervised protein language model which takes as input a set of MSA sequences.
 Source:MSA Transformer, Roshan Rao et al.
Source:MSA Transformer, Roshan Rao et al.
They used a shared (tied) row representation for all the rows so as to process the input MSA since there is a great structure overlap. It can be implemented by averaging the row head attention representations. This change is happening in the self-attention operation.
On the right, you can see the fundamental building block of their transformer encoder. Alternating row and column attention is called axial attention.
Conceptually, Alphafold2 uses a similar form of axial attention for MSA processing, although a bit more complex. Axial attention means that information is sequentially aggregated and routed in a row and a column level. The row-wise attention processes the relevant sequences separately, while the column attention combines residue information between different sequences of the MSA.
 Source: Axial-DeepLab: Stand-Alone Axial-Attention for Panoptic Segmentation
Source: Axial-DeepLab: Stand-Alone Axial-Attention for Panoptic Segmentation
This idea has been proposed in computer vision to deal with the quadratic complexity of self-attention.
AlphaFold2 core self-attention module: Invariant point attention
The core engineering on AlphaFold2 was the design of a transformer architecture that respects the particularities of the 3D domain and proteins. To this end, the DeepMind team proposed the Invariant Point Attention (IPA) module.
It is a form of attention that acts on a set of representations and is invariant under global Euclidean transformations (roto-translations). Again, this gives the model an easier time. For the record, to get a smooth landscape of roto-translations, they used quaternions instead of rotation/translation matrices.
 Invariant point attention.
Top blue arrays: Invariant Point Attention Module.
Middle, red arrays: modulation by the pair representation.
Bottom, green arrays: standard attention on abstract features.
Dimensions: r: residues, c: channels, h: heads, p: points. Source: AlphaFold2 supplementary material
Invariant point attention.
Top blue arrays: Invariant Point Attention Module.
Middle, red arrays: modulation by the pair representation.
Bottom, green arrays: standard attention on abstract features.
Dimensions: r: residues, c: channels, h: heads, p: points. Source: AlphaFold2 supplementary material
The depicted “pair representation” is computed based on the MSA. From the diagram we can infer that:
The single representation has two sets of which stand for queries, keys, values, so 6 in total.
The red square in the middle is the dot-product attention operation (before softmax).
Before the softmax operation two extra representations, coming from the backbone frames and pair representation, are added.
After the softmax-normalized attention weights, the information is routed based on 3 different value representations.
The 3 different outputs are aggregated and passed to the next layer.
Be sure to check our detailed article on vanilla self-attention to grasp a high-level overview of the core AlphaFold2 module. For more information on equivariance we suggest Fabian Fuchs & Justas Dauparas blogpost.
Conclusion: protein folding is still not solved
Before the official release of AlphaFold 2, an awesome open-source initiative to reproduce it started from EleutherAI. Their repository is under construction mostly by Phil Wang and Eric Alcaide. Meanwhile, there are many other researchers working on it. OpenFold2 is another attempt at replicating AlphaFold2. It divides the different segments that you need to solve for such a hard problem.
As a final word, we conclude with Dmitry Korkin’s awesome interview with Lex Fridman:
“AlphaFold 2 is amazing but protein folding is still not solved.” ~ Dmitry Korkin
This statement is made because real-life proteins are multi-domain, while the CASP completion is constrained to 1-2 protein domains.
We hope this article bridged many gaps between biology/bioinformatics and machine/deep learning. Feel free to share it on social media as a reward for our work. It is the best thing to help us reach the AI community.
Finally, you can find an overview of the AlphaFold2 paper:
Resources on AlphaFold2 and biology ML
[Official paper] Highly accurate protein structure prediction with AlphaFold
[Official notebook] - AlphaFold Colab
[Library] - Sidechainnet library
[Notebook] - Minimal version of AlphaFold2 (designed to work with a single sequence) with pre-trained weights from Deepmind created by @sokrypton
[Github repo] - An example of how the invariant point attention can be used in older CASP competitions by @lucidrains
[Blog] - AlphaFold 2 is here: what’s behind the structure prediction miracle by Oxford Protein Informatics Group
[Blog] - AlphaFold 2 & Equivariance by Justas Dauparas & Fabian Fuchs
[Notebook] - Training a CNN on random 5' UTR data along with hyper-parameter search
[Notebook] - CNN predictions of random 5' UTR growth rates
[Github repo] - Code from Deep Learning Of The Regulatory Grammar Of Yeast 5' Untranslated Regions from 500,000 Random Sequences
[Github repo] - Code from Sequence-to-function deep learning frameworks for engineered riboregulators
[Vid] - Visualizing quaternions (4d numbers) with stereographic projection - 3blue1brown
[Blog] - AlphaFold 2 & Equivariance
[Vid] - AlphaFold 2 is amazing but protein folding is still not solved | Dmitry Korkin and Lex Fridman
Cited as
@article{adaloglou2021biology,title = "Deep learning on computational biology and bioinformatics tutorial: from DNA to protein folding and alphafold2",author = "Adaloglou, Nikolas and Nikolados, Evangelos-Marios and Karagiannakos,Sergios",journal = "https://theaisummer.com/",year = "2021",howpublished = {https://github.com/The-AI-Summer/deep-learning-biology-alphafold},}

* Disclosure: Please note that some of the links above might be affiliate links, and at no additional cost to you, we will earn a commission if you decide to make a purchase after clicking through.